厚生労働科学研究費補助金 難治性疾患政策研究事業 小児腎領域の希少・難治性疾患群の全国診療・研究体制の構築
研究代表者 石倉健司
ロウ症候群とは、1)眼症状、2)中枢神経症状、3)腎症状の3つの症状を示す遺伝性の病気です。基本的には男の子に発症しますが、ごく稀に女の子にも発症すると言われています。

ロウ症候群の患者さんでは以下の3つの症状を認めます。
1) 眼症状
先天性白内障などの異常がみられます。先天性白内障は、生まれた時からほぼ全例に両側性に認められます。視力は大部分の患者さんが0.2以下と言われています。
2)
神経症状
認知機能の障害、精神運動発達遅滞、けいれん、行動異常、筋緊張低下などがみられます。
認知機能の障害や発達遅滞の程度には個人差がありますが、大半は軽度から中等度の障害となります。多くの患者さんで自立歩行が可能となるのは6-13歳と言われています。半数弱の患者さんにけいれん発作を認めますが、発作形式には個人差があります。自傷行為などの行動異常が加齢に伴い、約半数に発症すると言われています。
3)
腎症状
近位尿細管という部分が障害され、アミノ酸、ブドウ糖、電解質が再吸収できずに尿中に漏れる状態になります(ファンコニ症候群)。その結果、体重増加不良、くる病、代謝性アシドーシス(血液が酸性になること)などの症状が現れる場合があります(表1)。また、尿中のカルシウム排泄が増加し、その結果、約半数の患者さんに腎臓の石灰化が認められます。国内の検討では、30-40代で末期腎不全に至る患者さんが多い傾向にあります。
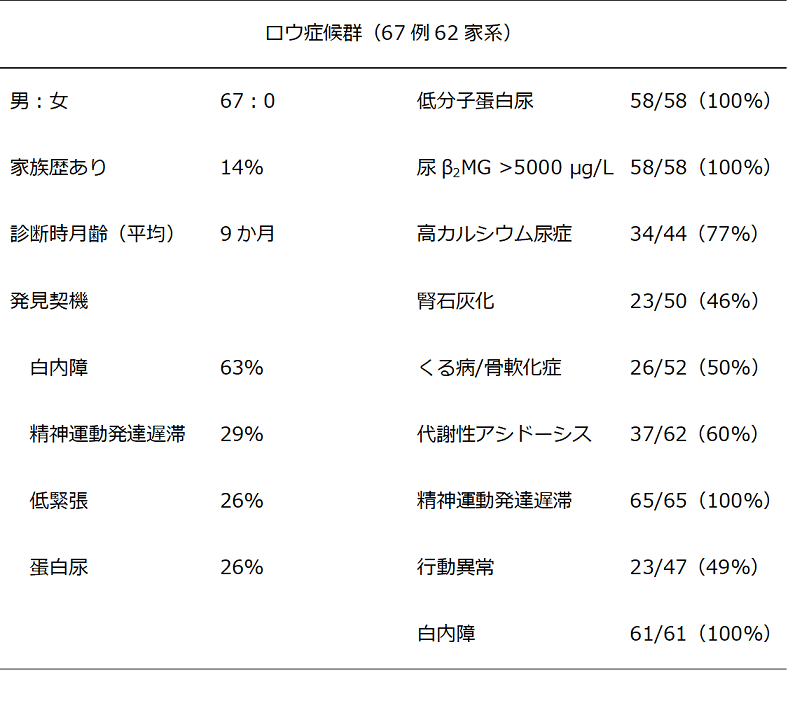
表1 国内のアンケート調査をもとにしたロウ症候群の症状
(平成27~28年度厚生労働科学研究(難治性疾患政策研究事業)「尿細管性蛋白尿を呈する遺伝性疾患の全国調査」より)
治療は、対症療法が中心となります。白内障に対しては手術が必要なことが多いです。けいれんに対しては抗けいれん薬を使用し、筋力低下に対しては理学療法を行います。腎不全に対しては、各種対症療法を行います。また、末期腎不全となり透析や腎移植を行った報告もあります。
男児10万人に数人の発症とされていますが、正確な発症頻度や患者数は不明です。
本症の原因として、X染色体上に責任遺伝子OCRLが同定されています。
X染色体劣性遺伝疾患であることがわかっています。
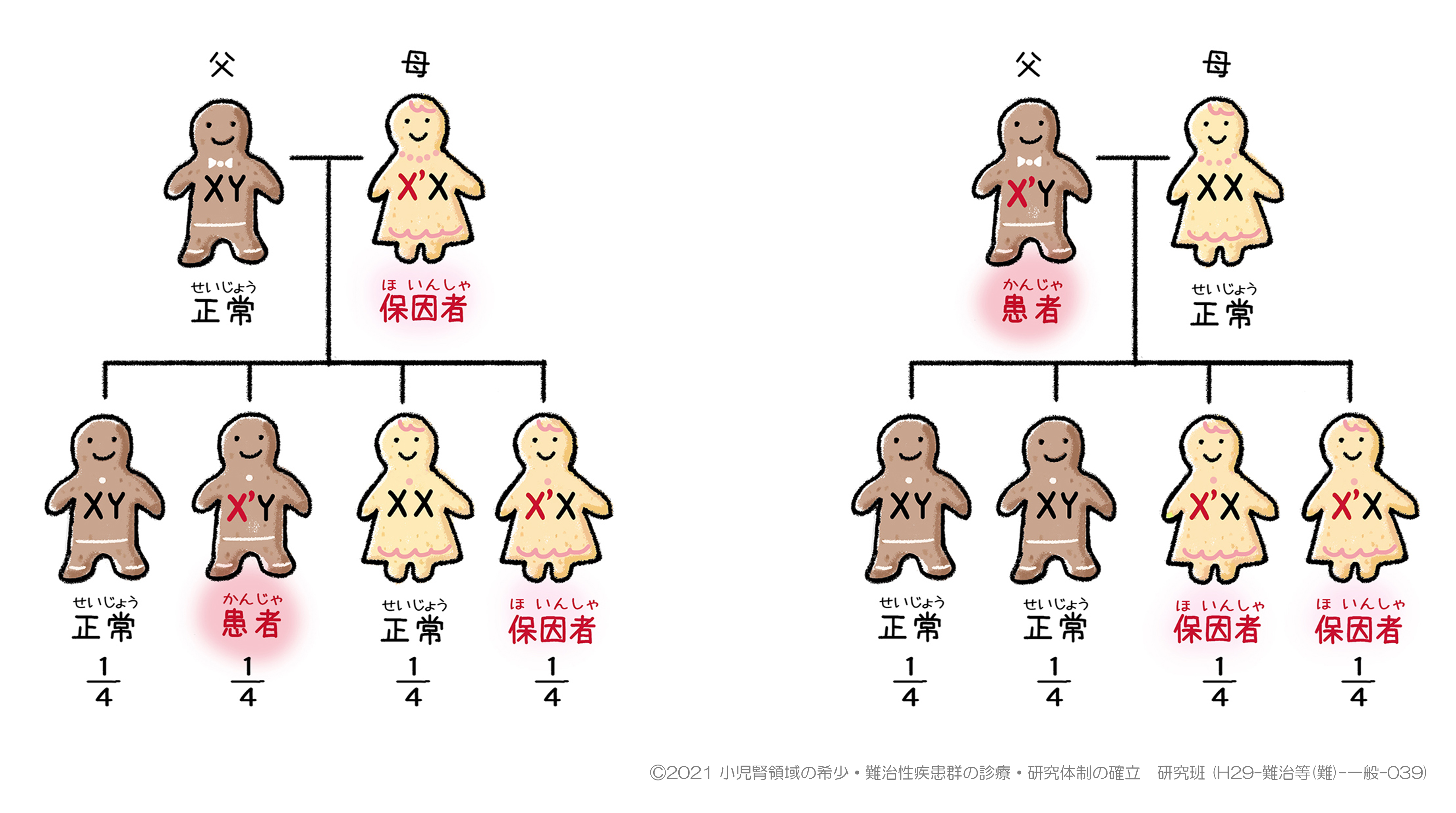
※解説
保因者とは、病気の原因となる遺伝子(図のX’ 染色体)を持っていますが、発症していない人を指します。
図1の場合、母は病気の原因となるX’
染色体を持っていますが、もう片方の染色体は正常(X染色体)なため、病気は発症せずに保因者となります。その子どものうち、一つしかX染色体を持たない男の子において、母のX’
染色体を受け継いだ場合は病気を発症します。また、女の子においては、仮にX’ 染色体を受け継いでも片方は正常なために、母と同様に保因者となります。
図2の場合、父は病気の原因となるX’
染色体を持っており、X染色体はこの一つだけであるため、病気を発症します。その子どものうち、男の子らは母から正常なX染色体を受け継ぐため正常です。一方で、女の子らは病気の原因となるX’
染色体を父から受け継ぐため、全員保因者となります。
治療は、対症療法が主体となります。腎機能障害は進行性で、30-40歳頃に末期腎不全に進行することが多いと言われています。
A
症状
1.先天性白内障
2.中枢神経症状(精神運動発達遅滞)
B
検査所見
1.尿中β2ミクログロブリン 5,000 µg/L以上
C
鑑別診断
Dent病、ミトコンドリア異常症、ガラクトース血症、遺伝性果糖不耐症、Fanconi-Bickel症候群
D
遺伝学的検査
1.OCRL遺伝子の変異
Definite:Aの2項目すべて+Bの1項目を満たしCの鑑別すべき疾患を除外し、Dを満たすもの
Probable:Aの2項目すべて+Bの1項目を満たしCの鑑別すべき疾患を除外したもの
表2 国内のアンケート調査をもとにしたLowe症候群の臨床症状の頻度
(平成27~28年度厚生労働科学研究(難治性疾患政策研究事業)「尿細管性蛋白尿を呈する遺伝性疾患の全国調査」より)


