Lowe syndrome is a hereditary disease characterized by three groups of symptoms: 1) eye manifestations, 2) central nervous manifestations, and 3) kidney manifestations. The disease is said to mostly occur in boys and very rarely in girls.

Patients with Lowe’s
syndrome present with three groups of symptoms listed below:
1)Eye manifestations
Abnormalities such as congenital cataract are observed. Congenital cataract is present bilaterally
from the time of birth in almost all patients. The majority of patients are reported to have a
vision of 0.2 or less.
2)Central nervous manifestations
Cognitive impairment, psychomotor retardation, convulsion, behavioral abnormalities, hypotonia, etc.
are present.
There are differences among individuals in the degree of cognitive impairment and developmental
retardation, but most commonly, the severity of the disorder is mild or moderate. It is said that
many patients become able to walk independently at the age of 6 to 13 years. Convulsive seizure
occurs in a little less than half of patients, but there are individual differences in the pattern
of seizures. Behavioral abnormalities such as self-harm behavior are reported to occur as aging in
about half of all patients.
3) Kidney manifestations
The dysfunction of a part of the kidneys called the proximal renal tubule causes the condition where
amino acids, glucose, and electrolytes that cannot be reabsorbed leak into the urine (Fanconi
syndrome). Consequently, symptoms such as failure to thrive, rickets, metabolic acidosis (the blood
becomes acidic) may appear (see table below). In addition, the excretion of calcium into urine
increases, and as a result, nephrocalcinosis is observed in about half of all patients. According to
an investigation in Japan, patients develop end-stage kidney disease most commonly in their 30s to
40s.
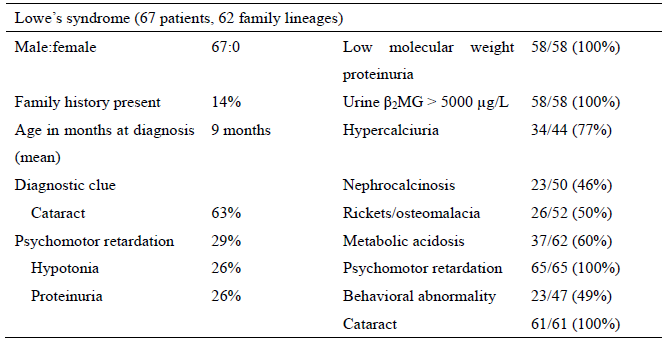
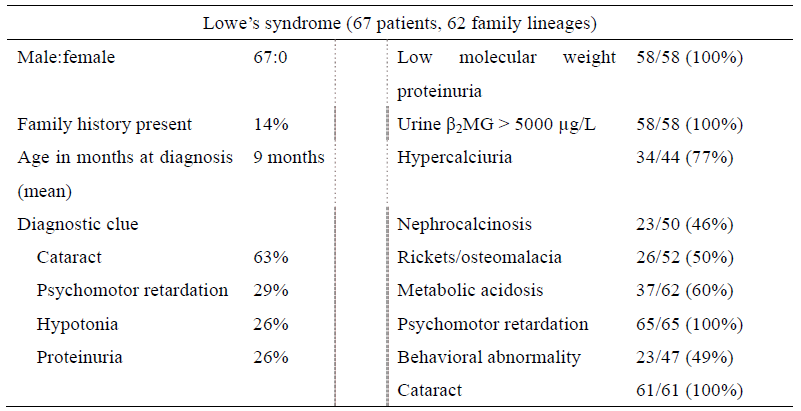
Table Symptoms of Lowe syndrome based on a questionnaire survey in Japan
(Cited from “A Nationwide Survey on Hereditary Diseases Presenting with Tubular Proteinuria. ”
Health and Labor Sciences Research Grants for FY 2015 to 2016 [Research Program on Rare and
Intractable Diseases])
Treatment is mainly symptomatic. Cataract often requires surgery. Convulsion is treated with anticonvulsant drugs, and muscle weakness is treated with physiotherapy. Various symptomatic treatments are given for kidney failure. There have also been some cases that progressed to end-stage kidney disease and required dialysis or kidney transplantation.
It is estimated that the disease occurs in a few out of 100,000 boys, but the accurate frequency and number of patients are unknown.
OCRL on chromosome X has been identified as the responsible gene causing this syndrome.
It is known to be an X chromosome recessive hereditary disease.
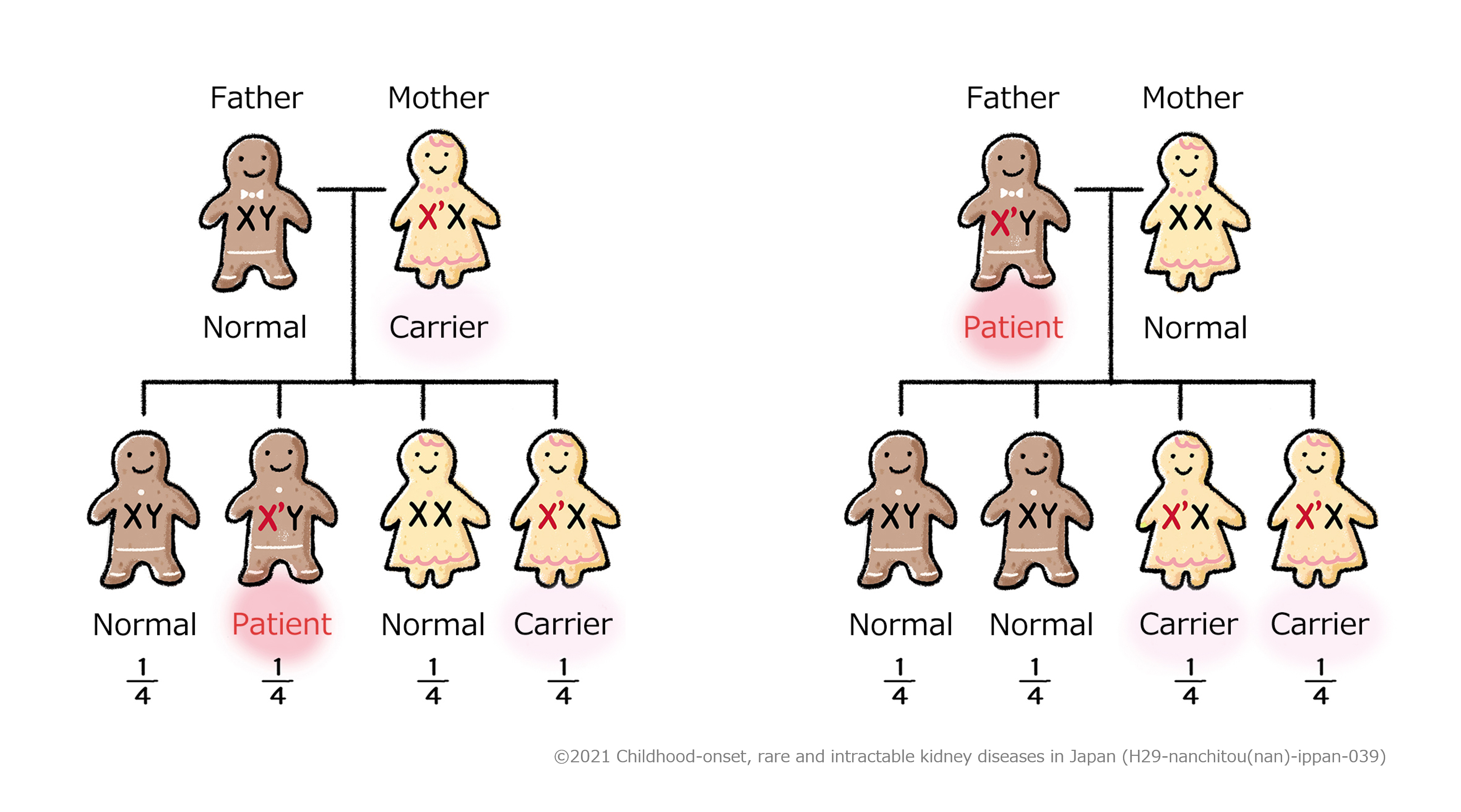
Explanation
Being a carrier means that a person carries a gene that can cause a disease (X’ chromosome in
the figure above) but does not develop the disease
In Figure 1, the mother carries an X’ chromosome causing the disease, but the other chromosome
(X chromosome) is normal, and so she is a carrier but does not develop the disease. Among her
children, if a son, who has only one X chromosome, inherits the X’ chromosome from her mother,
he develops the disease. If a daughter inherits the X’ chromosome from her mother, the other
chromosome is normal, so she becomes a carrier like her mother.
In Figure 2, the father has an X’ chromosome causing the disease, and this is the only X
chromosome he has, and so he develops the disease. Among his children, because sons inherit a
normal X chromosome from their mother, they are normal. On the other hand, girls inherit the X’
chromosome causing the disease from their father, and so they both become carriers.
Treatment is mainly symptomatic. Renal dysfunction is progressive, and patients are reported to progress to end-stage kidney disease most commonly in their 30s to 40s.
A Symptoms
1.Congenital cataract
2.Central nervous manifestations (psychomotor retardation)
B Laboratory findings
1.Urineβ2MG >5000 µg/L
C Differential diagnoses
Dent disease, mitochondrial disorders, galactosemia, hereditary fructose intolerance,
Fanconi-Bickel syndrome
D Genetic testing
1.Mutation in the OCRL gene
Definite:2 items of A + 1 item of B are met, differential diagnoses of C are excluded, and D is met.
Probable:2 items of A + 1 item of B are met, and differential diagnoses of C are excluded.
Table 1 Frequency of the clinical symptoms of Lowe syndrome based on a questionnaire survey in
Japan
(Cited from “A Nationwide Survey on Hereditary Diseases Presenting with Tubular Proteinuria.”
Health and Labor Sciences Research Grants for FY 2015 to 2016 [Research Program on Rare and
Intractable Diseases])