Galloway-Mowat syndrome is a syndrome that is characterized by three primary symptoms:
microcephaly, high-level proteinuria, and morphological defect of face (auricles, etc.) due to
developmental abnormalities of two organs, which are the head (cranial nerve) and kidneys. The disease
was named after English pediatric surgeons, Galloway and Mowat who reported the disease for the first time
in 1968.
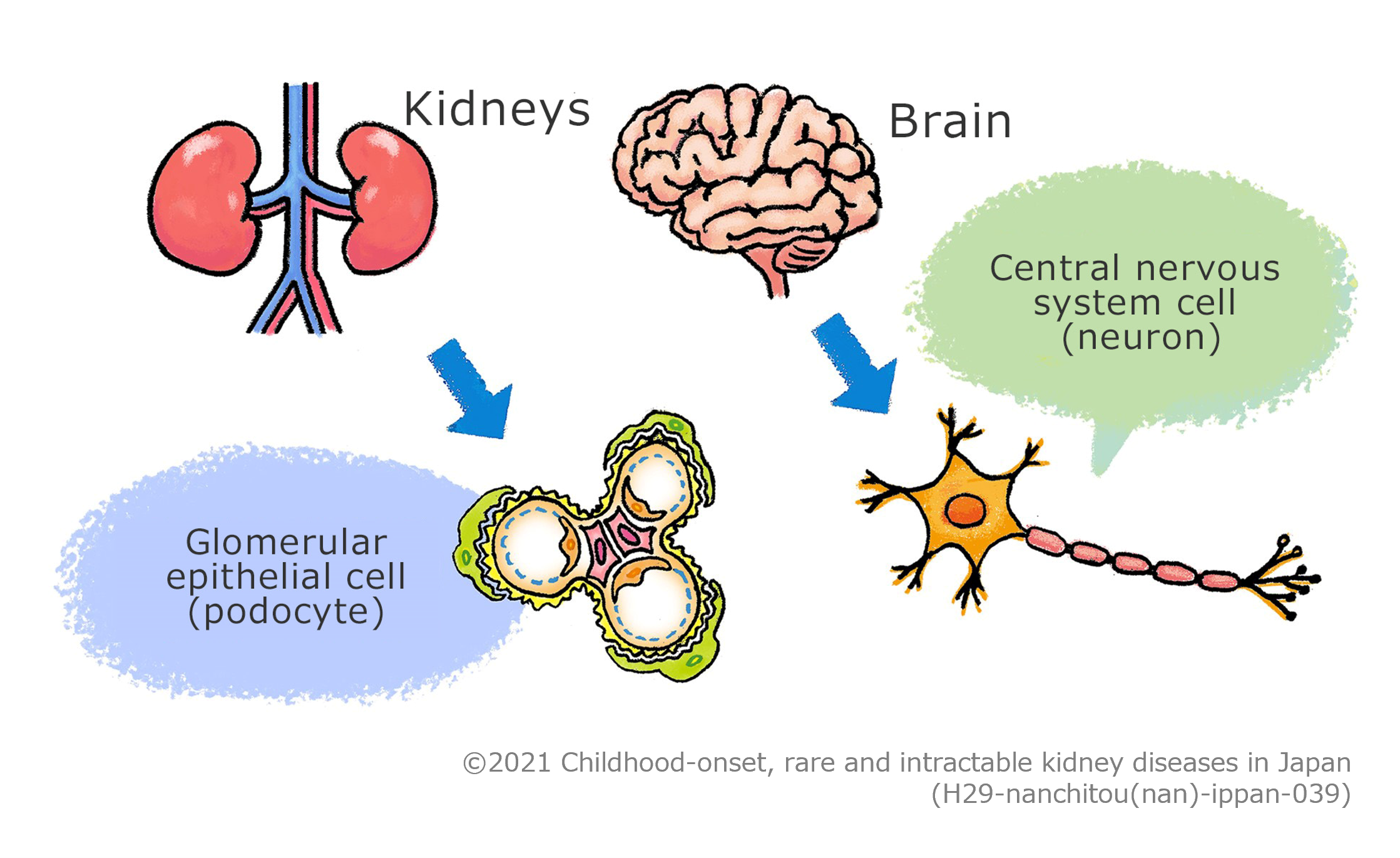
It is assumed that the disease is caused by cellular dysfunction that is shared by the glomerular
epithelial cells (podocytes) constituting the glomerulus, which is the filter of the kidneys and the nervous
cells (neurons) constituting the brain, and the dysfunction is suspected to cause abnormalities in the
process of development of the kidneys and the brain; however, definitive chromosomal aberrations or genetic
mutations that cause the disease have not been identified yet.
A tape measure is used to measure the distance from the glabella
(point on the frontal bone above the root of the nose) to the most posterior prominent point of the
occipital bone. Measurement should be read in millimeters.
※Measure the circumference of the head at the point just above both eyebrows anteriorly and the most
prominent point of the occipital bone posteriorly. Note that the tape should not be placed over the most
prominent part on the forehead.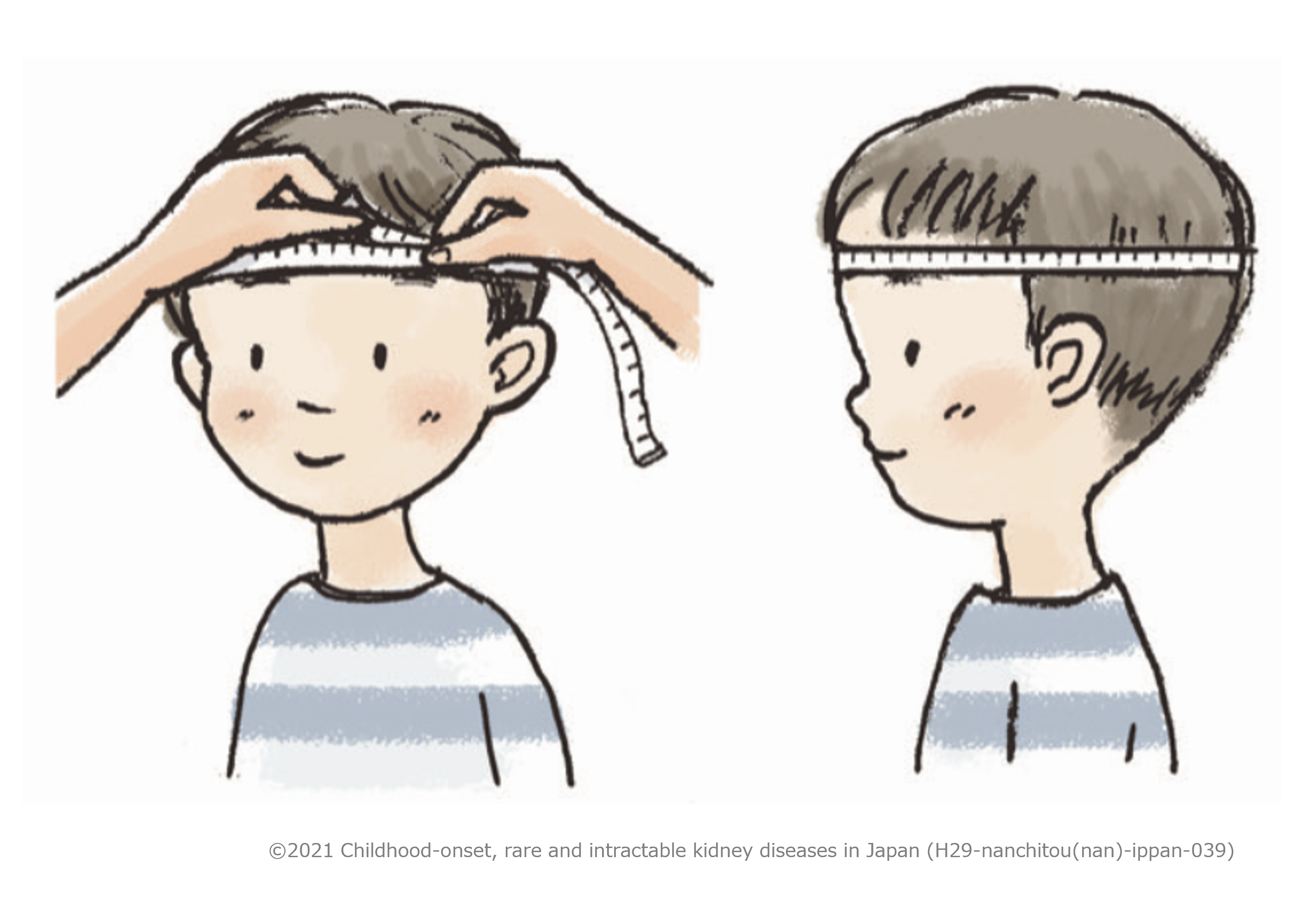
Percentile Curve of Head Circumference Measurement in Infants (2010
survey)

Reason for setting criteria different from that of Japan Intractable Diseases Information Center
Since causative genes are unknown, diagnostic criteria are critical. Nevertheless, there is no
internationally definitive diagnostic criteria, and the diagnostic criteria of the Japan Intractable
Diseases Information Center aim to identify all the patients with the disease without any omission. In
contrast to the criteria of the Japan Intractable Diseases Information Center, the present diagnostic
criteria have been established to accurately comprehend the patients who present significant symptoms of the
disease to identify causal genes in future. Therefore, the coverage of diagnosis has been narrowed down.
In many cases, microcephaly is associated with psychomotor retardation and refractory epilepsy. CT or MRI often reveals cortical dysplasia (gyrus abnormalities, white matter myelination deficiency) and structural aberration of the brain such as cerebellar hypoplasia.
The patients present with high-level proteinuria (urine
protein/creatinine ratio ≥ 1.0 g/gCr, or daily urine protein ≥ 1 g) where steroids are not
effective or are predicted not to be effective. In typical serious cases, massive proteinuria (nephrotic
syndrome) is seen by 3 months after birth. Nephropathy is a progressive disease and often leads to renal
failure; however, the renal failure tends to present with wide-ranging severity and progress to the end
stage between 3 and 10 years of age or older.
There are pediatric patients with mild nephropathy (proteinuria) or microcephaly (epilepsy or growth
retardation) who grow into adults with comparatively favorable courses. Meanwhile, there are other
patients who present with epileptic manifestation first and then develop nephrotic syndrome later. Focal
segmental glomerulosclerosis is often observed in renal biopsy tissue.
The disease is associated with ear abnormalities (low-set ears or floppy ears, etc.). It may also be associated with other facial morphological defects (narrow forehead, small jaw bone, high palate, wide-set eyes, etc.).
The patients may develop muscular hypotonia that may cause respiratory disorder and swallowing disorder. Complications of squint or hiatal hernia (a part of the stomach entrance slips through the diaphragm) may also be observed.
As there is no cure for this disease, treatment to alleviate the symptoms associated with the disease (symptomatic treatment) will be predominate. Nephropathy is progressive, and it should be treated as needed according to the disease stage: conservative phase (with renal disorder that does not yet need dialysis or renal transplantation), dialysis phase, and renal transplantation phase. Long-term medication is required for treatment of epilepsy.
In general, the symptoms are progressive. Many serious cases of early onset where the disease develops by 3 months after birth may progress to mental retardation or renal impairment due to epilepsy and die before 1-2 years of age. However, the degrees of renal impairment and neurological dysfunction vary among patients, and some patients develop the disease so slowly that they may grow up into adults with minimal interference of normal activity.
If the renal disorder is associated with Galloway-Mowat Syndrome, the only treatment that can be provided is to mitigate the symptoms associated with the disease (symptomatic treatment). However, there is a possibility that it is a complication of a disease that can be treated, so you should consult with a kidney diseases specialist.
The disease was named “Focal segmental glomerulosclerosis” looking at the kidneys histopathologically, without referring to the cause of the disease. To put it briefly, there are two types of the disease: one is caused by immune system disfunction and the other is caused by a genetically abnormal blood-filtering structure of the kidney. In the former cases, recurrence occurs frequently following renal transplantation, while recurrence does not occur in the latter. The renal lesion associated with Galloway-Mowat syndrome is the latter case; and therefore, basically you would not need to worry about recurrence.
It is difficult to answer because we do not have genetic information; however, considering that the disease is chiefly due to autosomal recessive inheritance, the sibling of a patient will develop the same disease in the ratio of 1/4. We cannot give you an accurate answer. Please consult with your doctor.

